1. Comprehensive genetic analysis of Epstein-Barr virus (EBV)-associated diseases
EBV infects >95% of the human population. After acute infection, EBV establishes a lifetime latent infection in B cells. The latent infection is not harmful to most people; however, this virus occasionally causes cancer or intractable diseases.
Chronic active EBV infection (CAEBV) is an intractable disease characterized by persistent and active EBV infection in T and/or NK cells. This disease is prevalent only in East Asia; however, including the reason that causes geographical bias, the molecular pathogenesis of this disease is unclear.
Next-generation sequencing (NGS)-based comprehensive analysis of human and EBV genomes in CAEBV identified the presence of somatic driver mutations in the human genome and large intragenic deletions in the viral genome.
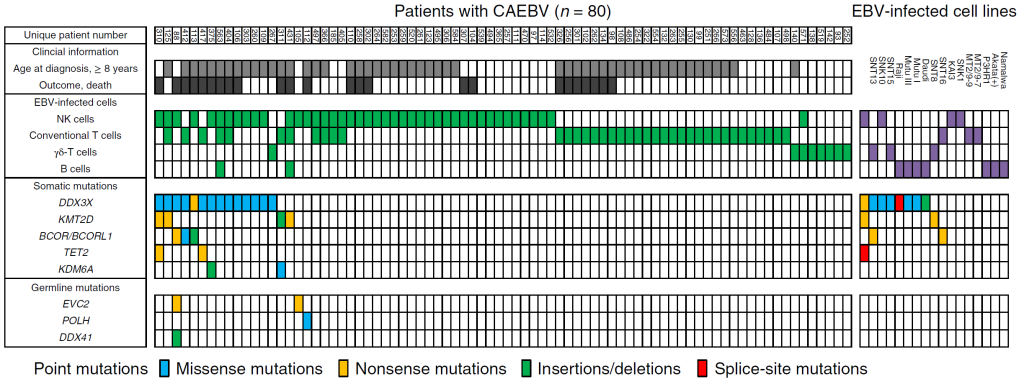
Figure 1. The mutational landscape of CAEBV
Each column indicates a patient with CAEBV (left) or an EBV-infected cell line (right). The rows contain clinical information, data on EBV-infected cells, and the somatic and germline mutations found. 20% of the patients carried somatic driver mutations such as DDX3X mutations in their EBV-infected cells.
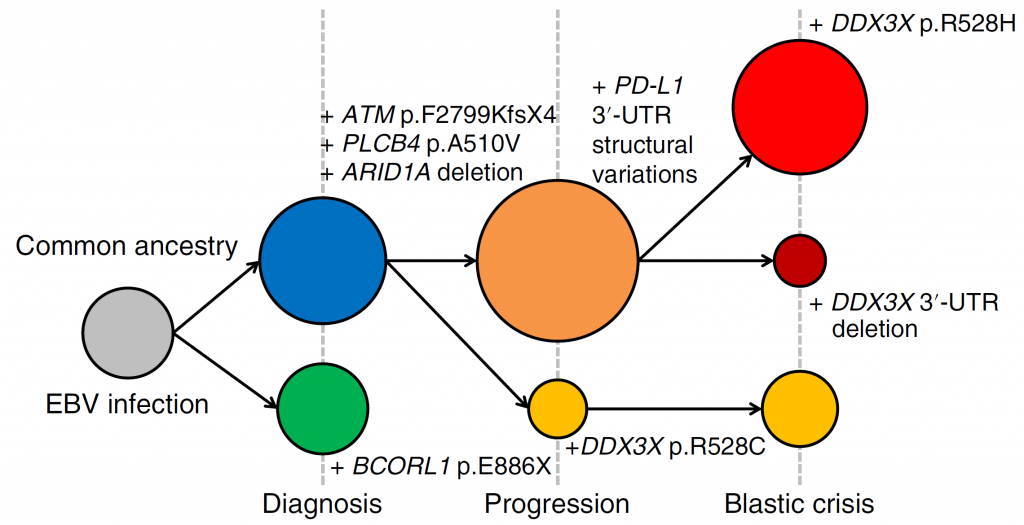
Figure 2. Clonal evolution of the EBV-infected cells in a patient with CAEBV
The EBV-infected cells in CAEBV acquired various driver mutations during the course of disease progression. Two separate clones were present at the time of diagnosis. At the time of progression, the clone with BCORL1 disappeared and the second clone acquired either additional PD-L1 3′ -UTR structural variations or a DDX3X p.R528C mutation. At the onset of blastic crisis, the clones with the additional PD-L1 3′-UTR structural variations acquired a DDX3X p.R528H mutation or a deletion affecting the DDX3X 3′ -UTR.
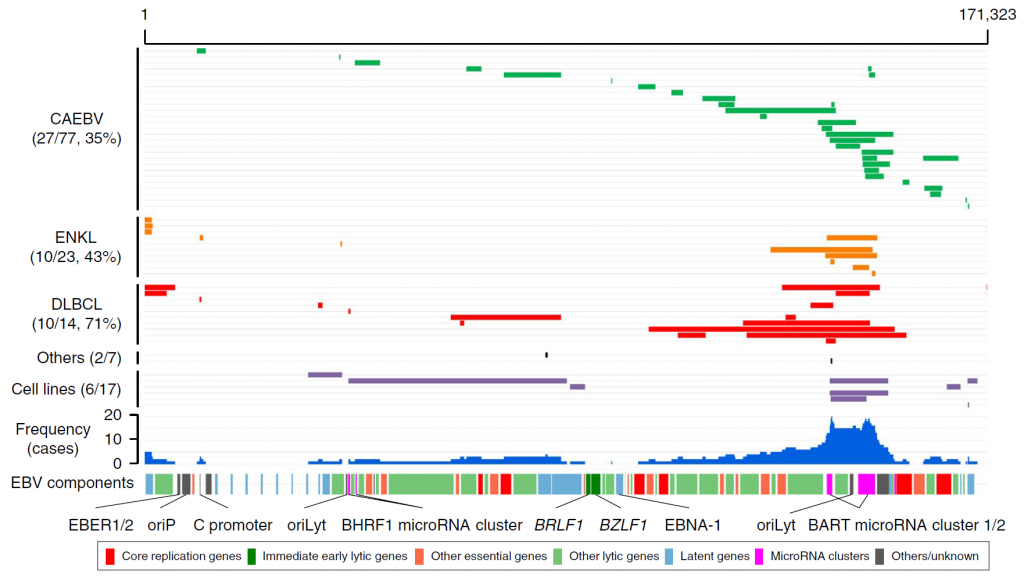
Figure 3. Intragenic deletions identified in EBV genomes
Each grey line indicates an EBV genome from a patient. The blue histograms indicate the frequency of the deletions in each genomic region. The locations of the components of the EBV genome are also shown. Large deletions of the viral genome were frequent across three diseases (CAEBV, extranodal NK/T-cell lymphoma, diffuse large B-cell lymphoma). BART microRNA clusters were the most frequently deleted components. Several viral genes that are essential for viral particle production were also frequently affected by deletions. Such deletions were considered to upregulate the lytic reactivation of EBV. oriP, replication origin used in latent infection; oriLyt, replication origin used in lytic infection.
This study revealed the malignant transformation of EBV-infected cells in CAEBV. This study also revealed frequent large deletions in EBV genomes of hematological malignancies. The lytic reactivation caused by the deletions is suggested to drive the malignant transformation of infected cells.
We are currently trying to expand this study by analyzing more EBV genomes from various diseases to find out the roles of the viral genome in malignancies.
References
1. Okuno Y et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nature Microbiology 2019;4(3):404-413.
2. Murata T et al. Oncogenesis of CAEBV revealed: Intragenic deletions in the viral genome and leaky expression of lytic genes. Reviews in Medical Virology 2020;30(2):e2095.
3. Mabuchi S et al. Role of Epstein-Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma. Cancers 2021;13(3).
2. Comprehensive genetic analysis of rare cancers and inherited diseases
2-1. Acute lymphoblastic leukemia (ALL)
ALL is the most frequent childhood neoplasm characterized by the most various somatic and germline mutations among cancers. The overall survival of B-cell ALL (B-ALL) is >90%; however, several kinds of gene mutations are negative predictors of prognosis.
To identify new gene mutations, we performed an NGS-based comprehensive genetic study of 59 children with relapsed B-ALL. As a result, we identified a novel rearrangement of MEF2D and BCL9 (MEF2D-BCL9 fusion gene) in 4 patients.
MEF2D-BCL9 was associated with the onset in adolescents, relapse (including relapse under treatment), and poor prognosis (0% survival in our cohort). The leukemic blasts with MEF2D-BCL9 were resistant to corticosteroids, which may explain the patients’ poor prognosis.
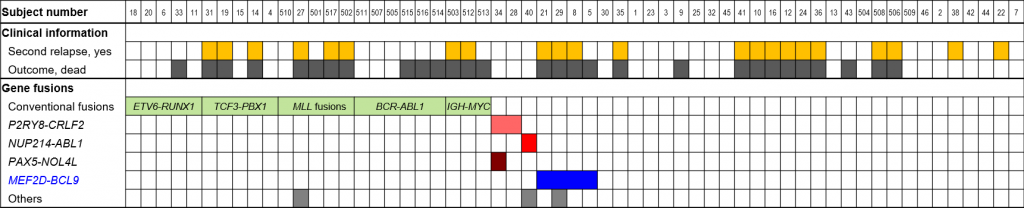
Figure 1. Mutational landscape of relapsed B-ALL
In addition to conventional fusion genes such as ETV6-RUNX1, novel MEF2D-BCL9 fusion was identified in 4 patients.
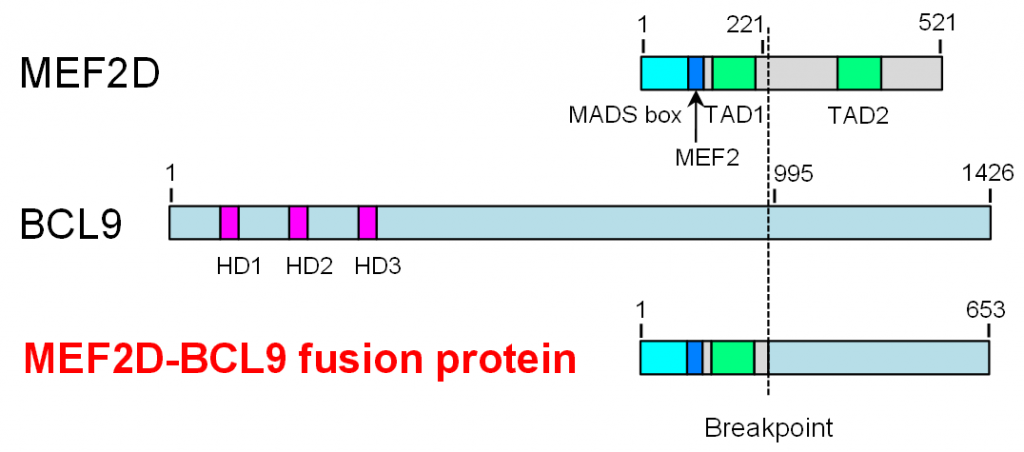
Figure 2. The structure of MEF2D-BCL9
An inversion of chromosome 1 caused an in-frame fusion of MEF2D and BCL9. The MEF2D-BCL9 fusion protein consisted of several functional domains derived from MEF2D and a C-terminal region of unknown function derived from BCL9.
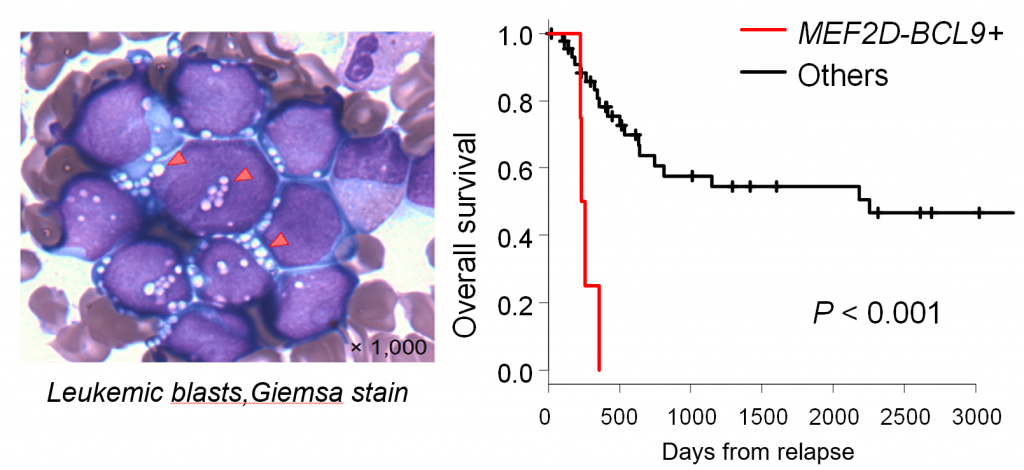
Figure 3. Clinical characteristics of B-ALL with MEF2D-BCL9
(Left) The Giemsa stain of leukemic blasts with MEF2D-BCL9. (Right) The prognosis of patients with MEF2D-BCL9.
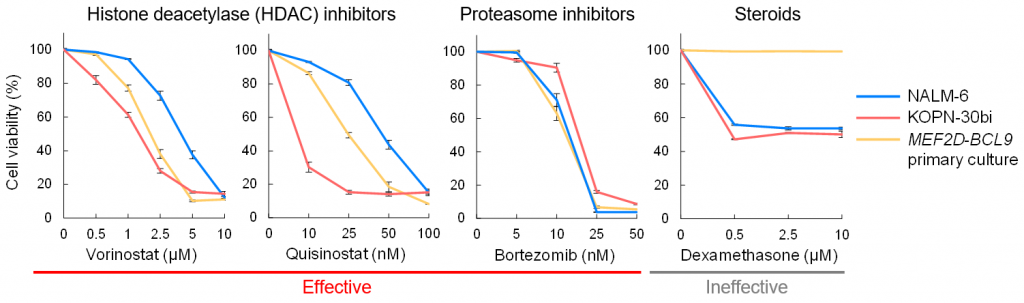
Figure 4. In vitro drug sensitivity of MEF2D-BCL9 blasts
A primary culture established from a patient with B-ALL carrying MEF2D-BCL9 was tested against several anti-cancer drugs.
This study identified the novel MEF2D-BCL9 in B-ALL. This fusion gene was associated with poor prognosis and corticosteroid resistance. Treatments using HDAC inhibitors may improve patients’ prognoses.
2-2. Juvenile myelomonocytic leukemia (JMML)
JMML is an intractable neoplasm of childhood. We performed a comprehensive multilayer genetic analysis of JMML using whole-exome sequencing, RNA sequencing, and array-based whole-DNA methylome analysis.
Whole-exome sequencing identified the mutually exclusive acquisition of the Ras pathway mutations. RNA sequencing identified three fusion genes that were not reported in JMML. All three fusions involved tyrosine kinases that can be targeted by tyrosine kinase inhibitors. Whole-DNA methylome analysis identified two subgroups of JMML with hugely different overall survival. The in-depth analysis identified a strong correlation among prognostic markers.
We treated a patient with JMML carrying the RANBP2-ALK fusion gene. Her disease was resistant to conventional chemotherapy. We utilized crizotinib and hematopoietic stem cell transplantation to achieve the long-term survival of the patient.
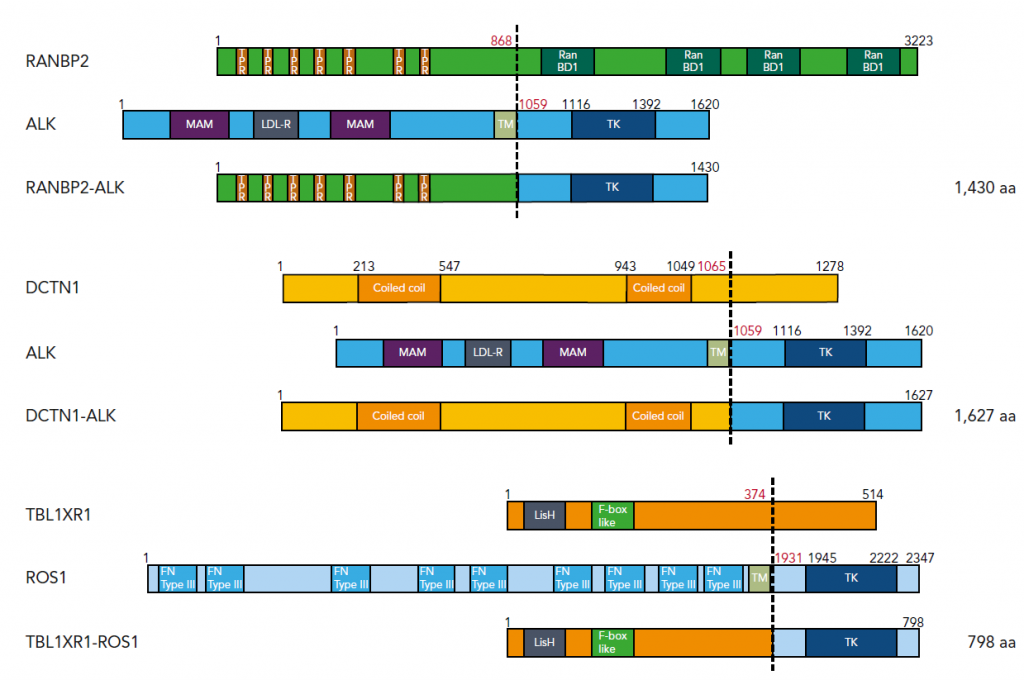
Figure 1. Three fusion genes identified in JMML
We identified fusion genes involving tyrosine kinases in JMML. ALK and ROS1 are within the same tyrosine kinase subfamily and can be targeted by ALK/ROS1 inhibitors such as crizotinib.
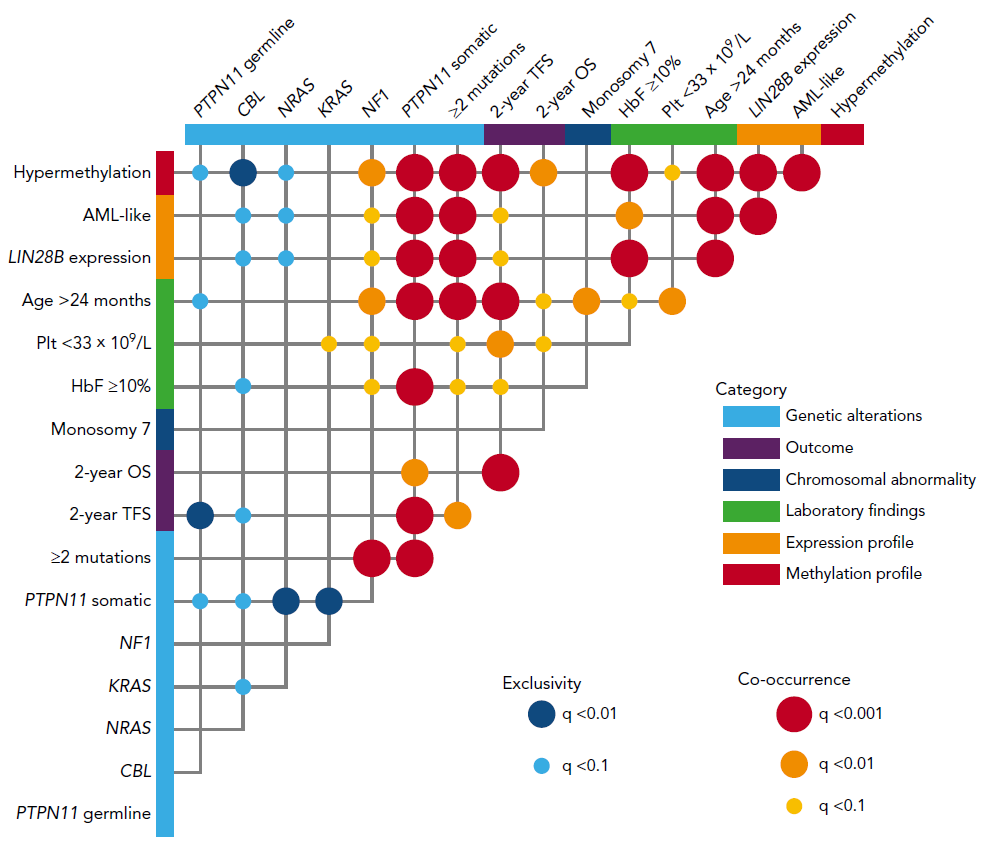
Figure 2. Correlation between prognosis markers in JMML
Poor prognosis markers were quite often co-occurrent within a patient.
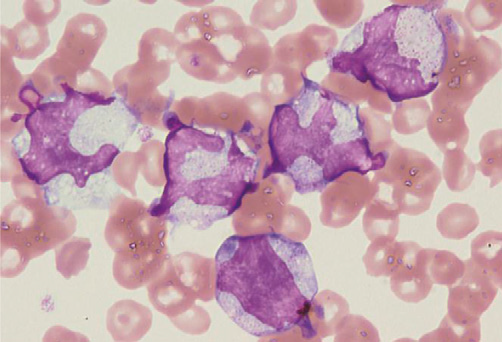
Figure 3. JMML blasts with RANBP2-ALK
Hugely distorted nuclei were characteristic.
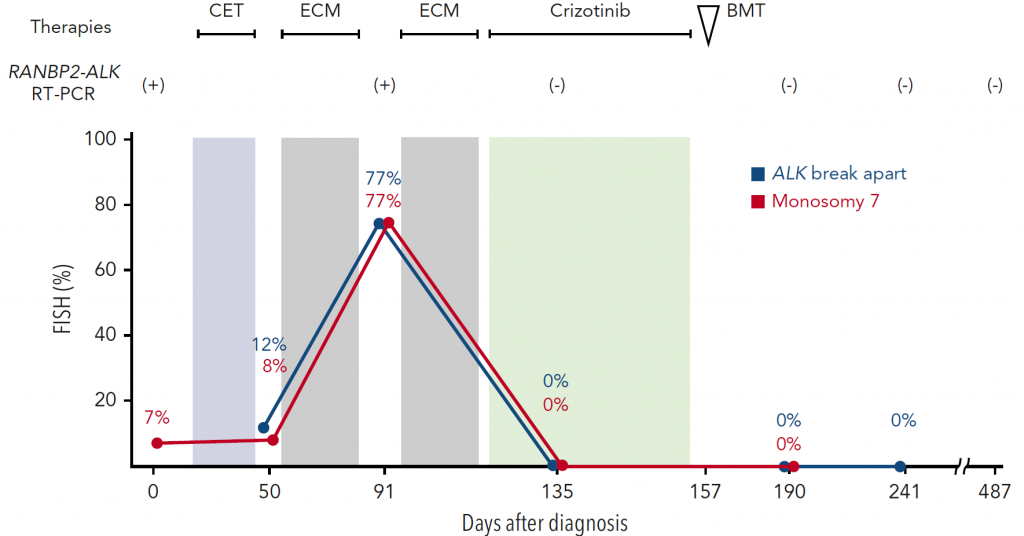
Figure 4. The treatment course of a JMML patient with RANBP2-ALK
The introduction of crizotinib resulted in the complete remission of her disease. Consolidation with bone marrow transplantation achieved long-term relapse-free survival.
Our study revealed the comprehensive genomic landscape, the precise risk prediction, and a fraction of patients who can be treated using ALK inhibitors. These findings contribute to stratified and targeted therapies of JMML.
2-3. Multicentric reticulohistiocytosis (MRH)
MRH is characterized by destructive arthritis that is somewhat similar to autoimmune arthritis in rheumatic arthritis (RA). One of the differences between arthritis in MRH and RA is the accumulation of histiocytes near affected joints in MRH.
The cause of this disease was unclear. Based on the similarity with RA, immunosuppressive therapies were utilized to treat patients with MRH. We performed a comprehensive genetic analysis of MRH to identify its molecular pathogenesis.
We analyzed two patients and identified somatic driver mutations in histiocytes of MRH. One was the MAP2K1 (MEK1) mutation that is frequently identified in histiocytic neoplasms such as Langerhans cell histiocytosis (LCH). The other was the KIF5B-FGFR1 tyrosine kinase fusion.
The presence of somatic driver mutations indicated the use of anti-cancer treatment regimens for MRH. We applied a kind of chemotherapy used for LCH to a patient with MRH and observed an objectively measurable response.
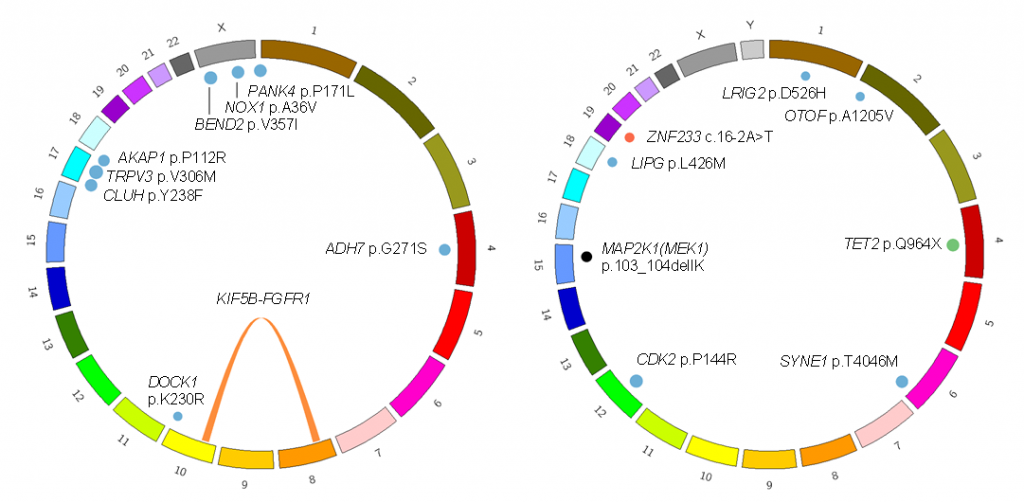
Figure 1. Somatic mutations identified in two patients with MRH
(Left) This patient carried the KIF5B-FGFR1 fusion gene and several passenger mutations. (Right) This patient carried the MAP2K1 p.103_104delIK and TET2 p.Q904* driver mutations.
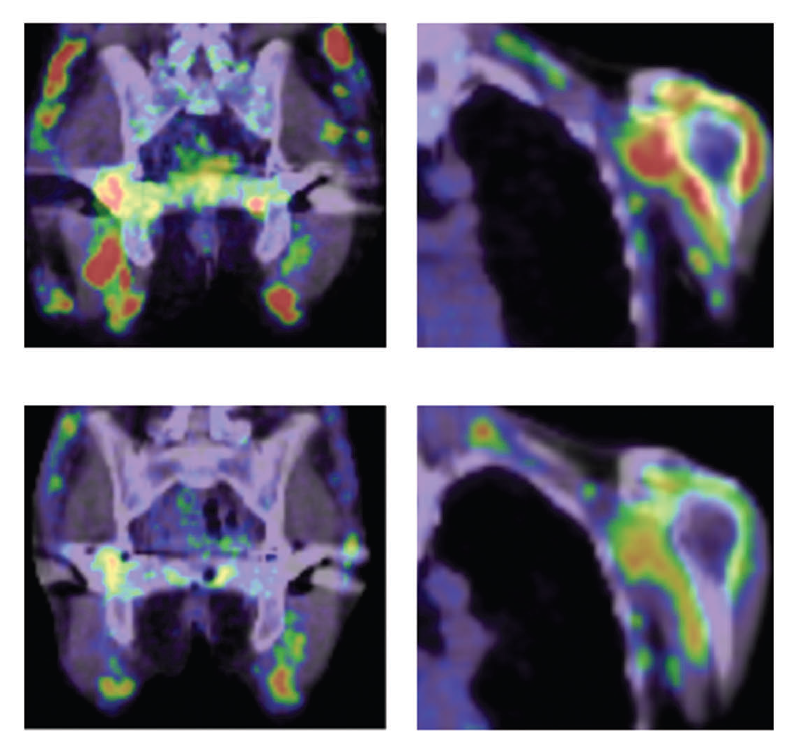
Figure 2. Treatment of an MRH patient with chemotherapy
(Upper) Before treatment. Red indicates inflammation of joints. (Lower) After treatment. No strong inflammation is indicated in joints.
Our analysis revealed a neoplastic nature of histiocytes in MRH. This finding contributed to the development of novel therapeutics using chemotherapies or may contribute to molecular target therapy using FGFR1 inhibitors.
2-4. Inherited macrothrombocytopenia (MTP)
MTP is characterized by giant platelets and mild to moderate bleeding tendency. We performed a comprehensive genetic study of families with autosomal dominant MTP. As a result, we identified mutations in ACTN1 (encoding alpha-actinin) in six families. Alpha-actinin cross-links actin filaments and its mutations disorganized cytoskeleton. Megakaryocytes with ACTN1 mutations produced large proplatelets in vitro.
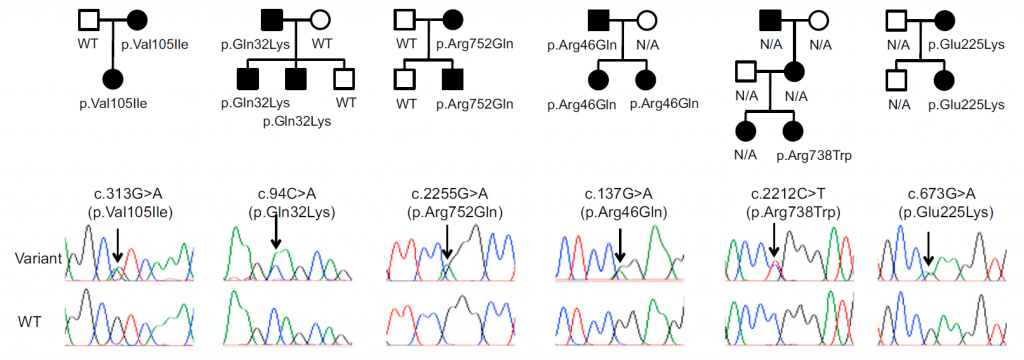
Figure 1. ACTN1 mutations found in families with autosomal dominant MTP
The identified ACTN1 variants showed strong co-segregation with clinical presentation.
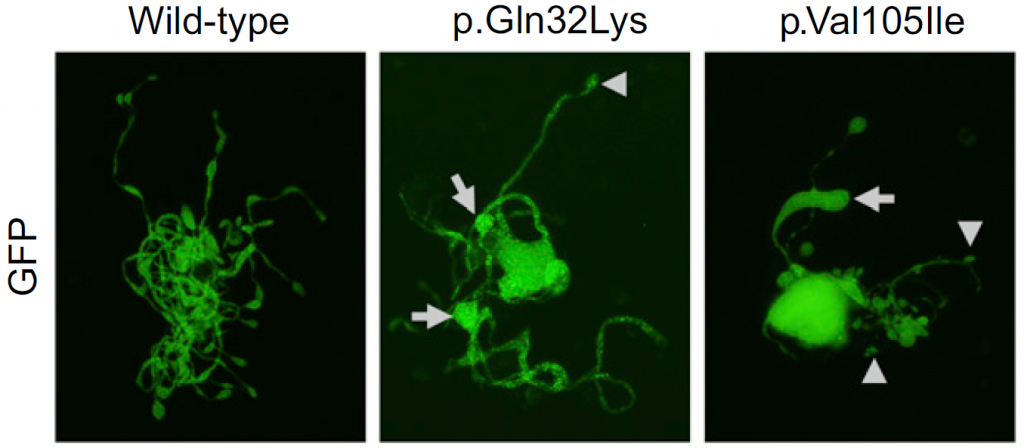
Figure 2. The effect of ACTN1 mutations on megakaryocytes in vitro
Mouse megakaryocytes were transfected with wildtype or mutated ACTN1. Large tips (resulting in large proplatelets) are indicated by arrows.
Currently, ACTN1 mutations are recognized as one of the most frequent causes of autosomal dominant MTP.
References
1. Suzuki K et al. MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. Journal of Clinical Oncology 2016;34(28):3451-3459.
2. Murakami N et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 2018;131(14):1576-1586.
3. Murakami N et al. Targetable driver mutations in multicentric reticulohistiocytosis. Haematologica 2020;105(2):e61-64.
4. Kunishima S et al. ACTN1 mutations cause congenital macrothrombocytopenia. American Journal of Human Genetics 2013;92(3):431-438.
3. Genetic diagnosis of patients with undiagnosed diseases
Inherited diseases are sometimes difficult to diagnose. Comprehensive genetic analyses such as whole-exome sequencing and targeted sequencing can identify causative mutations and establish genetic diagnoses. We performed comprehensive genetic studies of inherited bone marrow failure syndromes (IBMFS) and primary immunodeficiency (PID). Targeted sequencing could identify causative point mutations and copy number alterations in IBMFS. Targeted sequencing could also identify causative mutations in PID.
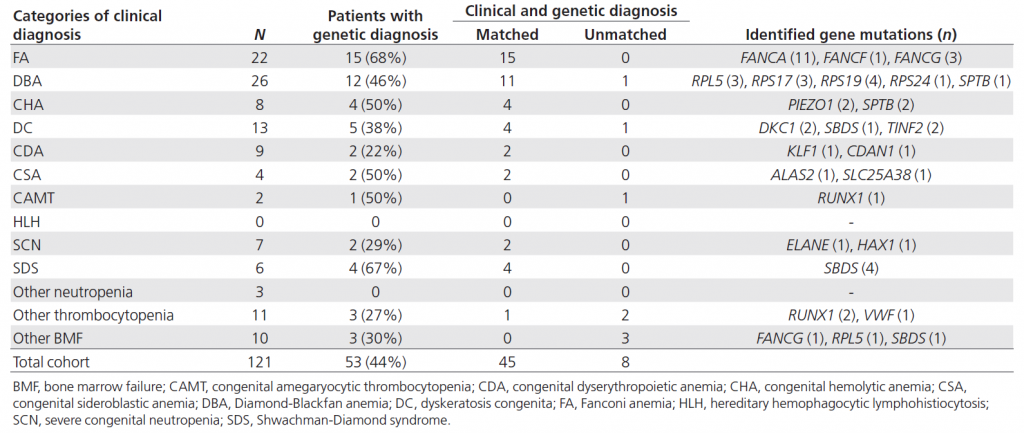
Table 1. Genetic diagnosis of IBMFS using targeted sequencing
Targeted sequencing could give genetic diagnoses in 44% of patients with IBMFS. Several genetic diagnoses were found to be discordant with clinical diagnoses.
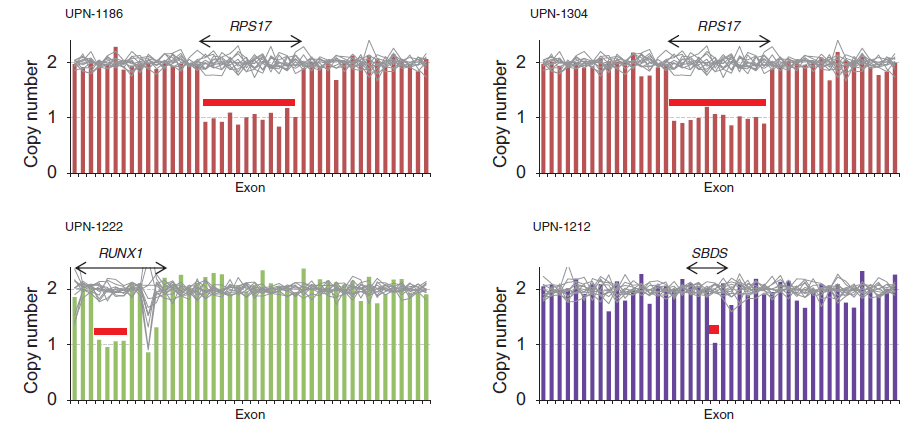
Figure 1. Copy number alterations identified by targeted sequencing
Heterozygous deletion of RPS17, RUNX1, and SBDS could be identified by targeted sequencing-based copy number analysis.
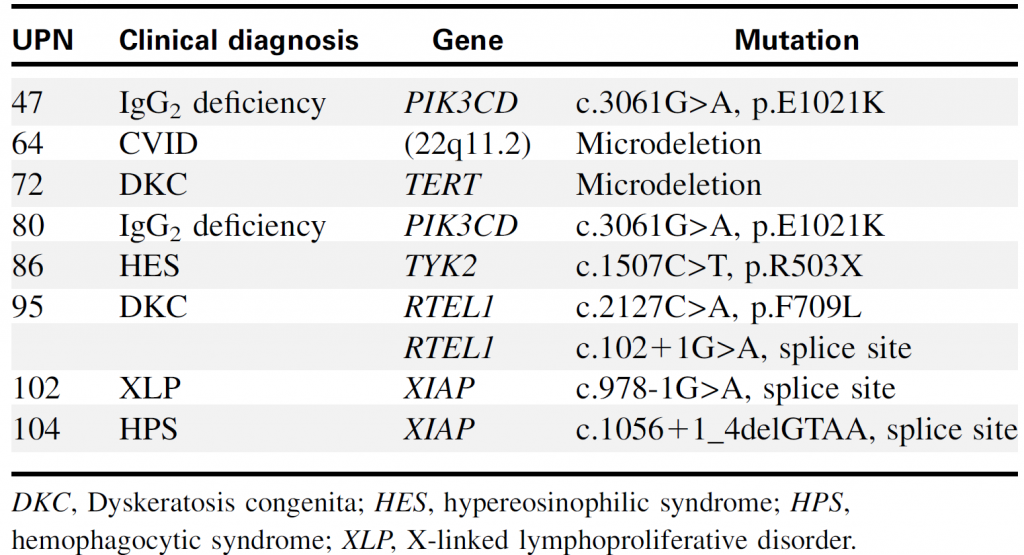
Table 2. Genetic diagnoses of PID identified by targeted sequencing
Targeted sequencing could identify point mutations and chromosomal copy number alterations (such as 22q11.2 microdeletion).
References
1. Muramatsu H et al. Clinical utility of next-generation sequencing for inherited bone marrow failure syndromes. Genetics in Medicine 2017;19(7):796-802.
2. Kojima D et al. Application of extensively targeted next-generation sequencing for the diagnosis of primary immunodeficiencies. Journal of Allergy and Clinical Immunology 2016;138(1):303-305.
4. Comprehensive detection of pathogens using next-generation sequencing
Next-generation sequencing (NGS) can determine the near-complete sequence of DNA or RNA samples. Therefore comprehensive detection of pathogens in clinical samples is theoretically possible by NGS. We performed a feasibility study using clinical samples. In the serum or cerebrospinal fluid samples of patients with acute encephalitis, we identified coxsackievirus A9, Mumps virus, and pepper mild mottle virus.
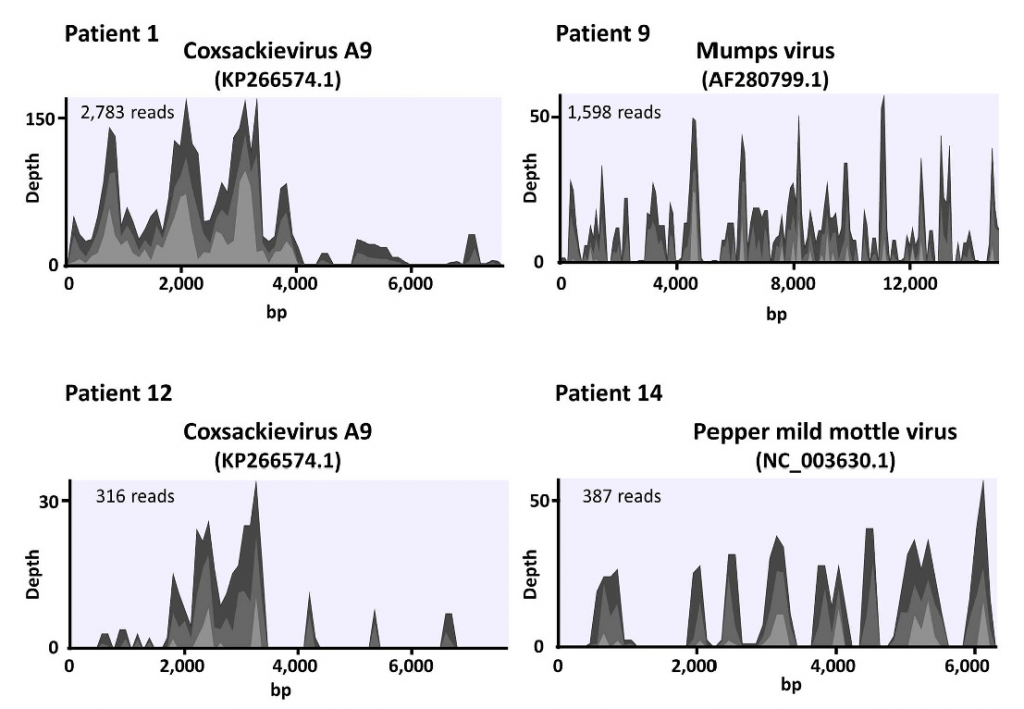
Several pathogens are difficult to identify using conventional methods such as bacterial culture. Detection of pathogens by NGS can complement current detection methods.
Reference
1. Kawada J et al. Identification of Viruses in Cases of Pediatric Acute Encephalitis and Encephalopathy Using Next-Generation Sequencing. Scientific Reports 2016;6:33452.
Publications
The full list of publications
https://www.ncbi.nlm.nih.gov/myncbi/1hqm7zm3FUf/bibliography/public/
Contact
Yusuke OKUNO, M.D., Ph.D
Professor
Department of Virology, Nagoya City University Graduate School of Medical Sciences